
PI’s: Gang Bao, Nael McCarty, & TJ Cradick
Cystic fibrosis (CF) is a common hereditary disease which affects the entire body, causing progressive disability and often, early death. CF is caused by a mutation in the CFTR (cystic fibrosis transmembrane conductance regulator) gene. The most common mutation, ΔF508, is a deletion (Δ) of three nucleotides that results in a loss of the amino acid phenylalanine (F) at the 508th (508) position on the protein. This mutation accounts for two-thirds of CF cases worldwide and 90 percent of cases in the US. Although most people without CF have two working copies (alleles) of the CFTR gene, only one is needed to prevent cystic fibrosis. CF develops when neither allele can produce a functional CFTR protein. Thus, CF is considered an autosomal recessive disease. While CF is a multi-organ disease, with a plethora of functional defects, most of the morbidity and mortality associated with CF results from the progressive loss of lung function. CF lung disease is due to chronic inflammation, persistent infection, impaired mucociliary clearance, and susceptibility to damage from oxidative stress.
Although intravenous, inhaled, and oral antibiotics can be used to treat chronic and acute infections, and mechanical devices and inhalation medications can be used to alter and clear the thickened mucus, these therapies can be extremely time-consuming for the patient and are of limited efficacy. In addition, gene therapy approaches have been developed that aim to introduce normal CFTR to airway epithelial cells. There are two CFTR gene introduction mechanisms involved, first the use of a viral vector (adenovirus, adeno-associated virus, or retro virus) and secondly the use of liposomes. However there are everal problems associated with these methods including efficiency (liposomes generate insufficient protein) and delivery (virus provokes an immune response).
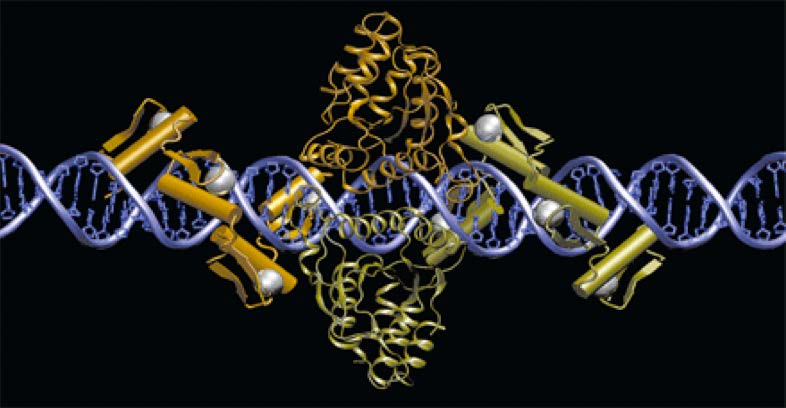
We propose to develop a paradigm-shifting approach to treat cystic fibrosis using a gene correction approach based on zinc finger nuclease (ZFN) and TAL effector nuclease (TALEN). As shown in Fig. 1, in this approach, two engineered nucleases (e.g., ZFN1 and ZFN2) bind to a specific region of the CFTR gene near
the mutation (e.g., ΔF508), generate a DNA double-strand break (DSB), and trigger the homologous recombination (HR) pathway for DNA repair. By supplying the correct template, the HR repair process corrects the mutation, thus restoring the normal function of the gene. We will establish the proof-of-concept of this novel methodology by performing studies with the following specific aims:
Aim 1. Design ZFNs and TALENs for generating a DNA DSB near the ΔF508 mutation in the CFTR gene and test in vitro. We will perform studies to design and test ZFNs/TALENs that will generate a DSB near the ΔF508 mutation in the CFTR gene. We will express the nucleases in vitro and determine their binding specificity and cutting efficiency.
Aim 2. Determine targeting specificity, off-target cleavage and levels of gene correction in cells. We will co-express nucleases and model substrates to measure the levels of DSB in cultured cells. This will allow measuring the rate of cleavage and repair at the specific target sequence, as well as non-specific offtarget locations. We will also deliver donor DNAs into cells to quantify and optimize the gene correction
efficiency.
The proposed research takes advantage of the expertise in Zinc Finger Nucleases design and gene correction that exists at the Nanomedicine Development Center Georgia Tech (Bao), and the expertise of researchers in the Center for Cystic Fibrosis Research in molecular analysis of CFTR function (McCarty), as well as the availability of mouse models through the Center for CF Research. Funding of this proposal via the pilot project program will promote sustainable interactions and collaborations, and lead to the development of preliminary data that are critical to the subsequent submission of this project as an NIH R01 proposal, or a proposal to the Cystic Fibrosis Foundation.