
PI’s: Gang Bao, Wilfried Rossol, & Han Phan
Spinal muscular atrophy (SMA) is a lethal childhood disease that is caused by low levels of the survival of motor neuron (SMN) protein. SMA is the leading genetic cause of death in infants, with an incidence of about 1 in every 6000 newborns. In the USA alone more than 25,000 people are believed to suffer from SMA, and currently there is no cure for SMA or treatment to stop its progression. To address this need we propose to develop engineered nucleases for genome editing to permanently restore SMN expression, and to optimize their delivery into motor neurons.
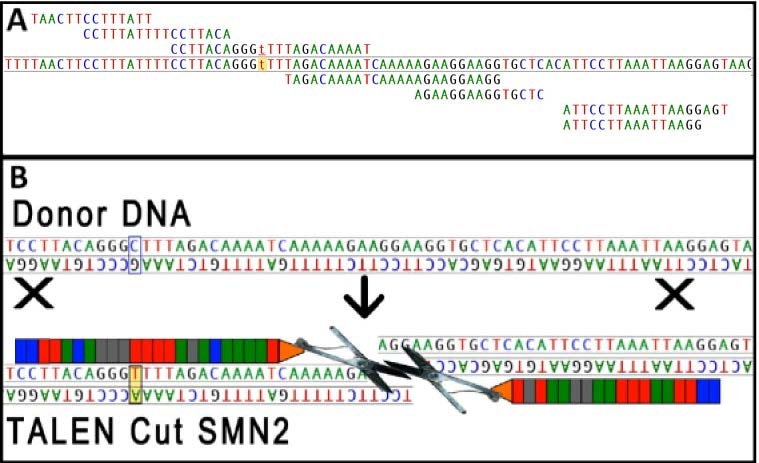
SMA is caused by homozygous deletions or mutations in the SMN1 gene encoding the survival motor neuron protein (SMN), which leads to a very specific degeneration of motor neurons in the anterior horn of the spinal cord. Humans also possess a second copy of the gene, SMN2, which encodes the same proteins. A single nucleotide change in a splice site in exon 7 results in aberrant skipping of exon 7 in approximately 90% of the SMN2 transcripts, encoding a truncated and unstable protein. While patients lack SMN1, they still retain at least one copy of SMN2.
The existence of the SMN2 gene in all SMA patients offers a unique therapeutic point of intervention. Results from SMA mouse models show that postnatal delivery of oligonucleotides that restore SMN2 splicing via intracerebroventricular injection, or viral delivery of SMN expression constructs can rescue the spinal muscular atrophy disease phenotype.
The objective of this proposed pilot project is to develop and optimize engineered nucleases and efficient delivery methods to correct the mutation in SMN2 locus as a novel approach for treating SMA. Transcription Activator-Like Effector Nucleases (TALENs) are engineered restriction enzymes generated by fusing the TAL effector DNA binding domain to a DNA cleavage domain. Our gene correction approach is based on the hypothesis that a pair of TALENs binds to the target DNA sequence, inducing a double-strand break and triggering homologous recombination (HR). When the exogenous DNA template is used to repair the break by HR, the mutation in the target gene is eliminated. This proposed innovative approach for treating SMA is based on a synergy between expertise in the Rossoll lab in SMA animal models and primary motor neurons, and that in the Bao lab in the design, validation and optimization of engineered nucleases.
We propose a systematic approach to optimize TALEN constructs using reporter cell lines and cultured SMA patient fibroblasts, and to evaluate the viral-based methods for delivering TALENs and templates into primary spinal cord motor neurons from SMA mouse models and stem cell-derived motor neurons, with the following specific aims:
Specific Aim 1: Design and optimize engineered nucleases that efficiently target the SMN2 locus and enhance SMN expression. We will design TALENs specific for the SMN2 locus and evaluate their specificity using in vitro cleavage assays. Functional TALEN pairs will then be optimized for genome editing of the SMN2 locus first in cell lines that carry SMN2-luciferase reporters. We will test the efficiency of TALENs targeted to the human SMN2 locus in patient fibroblasts and motor neurons cultured from SMA mouse models that carry the human SMN2 gene. The level of specific cleavage and possible off-target evens will be detected using bioinformatics and mismatch-specific DNA endonuclease assays. Expression of SMN will be quantified with immunostaining and western blots.
Specific Aim 2: Optimize delivery of TALEN constructs into motor neurons via transduction with adeno-associated virus (AAV). We will clone TALENs into AAV constructs and test them for delivery into cultured primary motor neurons and stem cell-derived motor neurons. In this aim, we will use AAV9 vectors from Dr. Foust's lab, which have been shown to transduce the majority of motor neurons when injected intravenously into neonatal mice. We will monitor delivery by measuring SMN expression and successful genome editing in target cells.
The proposed experiments have important advantages over current treatment strategies. Current efforts to develop treatments for SMA require constant treatment to up-regulate SMN protein levels. We believe that our novel approach has the potential to provide a more permanent solution. At the end of the proposed study, we will have identified and characterized engineered nuclease constructs that restore SMN expression in SMA motor neurons. TALENs that restore SMN expression in cultured motor neurons and in SMA animal models are likely to work in patients. Subsequent studies after the one-year pilot include the use of an intermediate SMA mouse model to test the efficacy of viral transduction and its effect on motor function, neuromuscular physiology and life span in vivo. Our proposed pilot project thus has the potential to establish the proof-of-concept of a novel treatment option for future clinical trials for SMA therapeutics.